*Raimondi, Daniele, Antoine Passemiers, Nora Verplaetse, Massimiliano Corso, Ángel Ferrero-Serrano, Nelson Nazzicari, Filippo Biscarini, Piero Fariselli, and Yves Moreau. "Biologically meaningful genome interpretation models to address data underdetermination for the leaf and seed ionome prediction in Arabidopsis thaliana." Scientific reports 14, no. 1 (2024): 13188.
*Gavalda-Garcia, J., Bickel, D., Roca-Martinez, J., Raimondi, D., Orlando, G., & Vranken, W. (2024). Data-driven probabilistic definition of the low energy conformational states of protein residues. NAR genomics and bioinformatics, 6(3), lqae082.
* Passemiers, A., Tuveri, S., Sudhakaran, D., Jatsenko, T., Laga, T., Punie, K., ... & Vermeesch, J. R. (2024). MetDecode: methylation-based deconvolution of cell-free DNA for noninvasive multi-cancer typing. Bioinformatics, 40(9), btae522.
*Nourisa, J., Passemiers, A., Shakeri, F., Omidi, M., Helmholz, H., Raimondi, D., ... & Zeller-Plumhoff, B. (2024). Gene regulatory network analysis identifies MYL1, MDH2, GLS, and TRIM28 as the principal proteins in the response of mesenchymal stem cells to Mg2+ ions. Computational and structural biotechnology journal, 23, 1773-1785.
*Passemiers, A., Folco, P., Raimondi, D., Birolo, G., Moreau, Y., & Fariselli, P. (2024). A quantitative benchmark of neural network feature selection methods for detecting nonlinear signals. Scientific Reports, 14(1), 31180.
* D. Raimondi, H. Chizari, N. Verplaetse, B.S. Löscher, A. Franke, Y. Moreau. “Genome interpretation in a federated learning context allows the multi-center exome-based risk prediction of Crohn’s disease patients” Scientific Reports 13 (1), 19449 (2023)
* N. Verplaetse, A. Passemiers, A. Arany, Y Moreau, D. Raimondi. “Large sample size and nonlinear sparse models outline epistatic effects in inflammatory bowel disease”. Genome Biology 24 (1), 224 (2023)
* Mazzone, Eugenio, Yves Moreau, Piero Fariselli, and Daniele Raimondi. "Nonlinear data fusion over Entity–Relation graphs for Drug–Target Interaction prediction." Bioinformatics 39, no. 6 (2023): tad348.
* Raimondi, Daniele, Francesco Codicè, Gabriele Orlando, Joost Schymkowitz, Frederic Rousseau, and Yves Moreau. "HPMPdb: A machine learning-ready database of protein molecular phenotypes associated to human missense variants." Current Research in Structural Biology 4 (2022): 167-174.
* Orlando, Gabriele, Daniele Raimondi, Francesco Codice, Francesco Tabaro, and Wim Vranken. Prediction of disordered regions in proteins with recurrent neural networks and protein dynamics. Journal of Molecular Biology 434, no. 12 (2022): 167579.
* Passemiers, Antoine, Yves Moreau, and Daniele Raimondi. Fast and accurate inference of gene regulatory networks through robust precision matrix estimation. Bioinformatics 38, no. 10 (2022): 2802-2809.
* G. Orlando, D. Raimondi, R. Duran-Romana, Y. Moreau, J. Schymkowitz, F. Rousseau, PyUUL: an interface between biological structures and deep learning algorithms, Nature Communications, 13 (1), 961 (2022)
* D. Raimondi, M. Corso, P. Fariselli, Y. Moreau, From genotype to phenotype in Arabidopsis thaliana: in-silico genome interpretation predicts 288 phenotypes from sequencing data, Nucleic Acids Research, 2021; gkab1099, https://doi.org/10.1093/nar/gkab1099 2022
* D. Raimondi, A. Passemiers, P. Fariselli, and Y. Moreau. Current cancer driver variant predictors learn to recognize driver genes instead of functional variants. BMC Biology, 19(1), 2021.
* L.P. Kagami, G. Orlando, D. Raimondi, F. Ancien, B. Dixit, J. Gavaldá-Garciá, P. Ramasamy, J. Roca-Martı́ nez, K. Tzavella, and W. Vranken. B2btools: Online predictions for protein biophysical features and their conservation. Nucleic Acids Research, 49(W1):W52–W59, 2021.* M. Necci, et al., Critical assessment of protein intrinsic disorder prediction. Nature Methods, 18(5):472–481, 2021.
* D. Raimondi, J. Simm, A. Arany, Y. Moreau, A novel method for data fusion over entity-relation graphs and its application to protein–protein interaction prediction, Bioinformatics, Volume 37, Issue 16, 15 August 2021, Pages 2275–2281, https://doi.org/10.1093/bioinformatics/btab092
* D. Raimondi, G. Orlando, E. Michiels, D. Pakravan, A. Bratek-Skicki, L. Van Den Bosch, Y. Moreau, F. Rousseau, J. Schymkowitz, In silico prediction of in vitro protein liquid–liquid phase separation experiments outcomes with multi-head neural attention, Bioinformatics, Volume 37, Issue 20, 15 October 2021, Pages 3473–3479, https://doi.org/10.1093/bioinformatics/btab350
* L.M. Peeters, et al., Covid-19 in people with multiple sclerosis: A global data sharing initiative. Multiple Sclerosis Journal, 26(10):1157–1162, 2020.
* G. Orlando, D. Raimondi, L. P. Kagami, W. F Vranken, ShiftCrypt: a web server to understand and biophysically align proteins through their NMR chemical shift values, Nucleic Acids Research, Volume 48, Issue W1, 02 July 2020, Pages W36–W40, https://doi.org/10.1093/nar/gkaa391
* G. Orlando, A. Silva, S. MacEdo-Ribeiro, D. Raimondi, and W. Vranken. Accurate prediction of protein beta-aggregation with generalized statistical potentials. Bioinformatics, 36(7):2076–2081, 2020.
* G. Buroni, Y.-A. Le Borgne, G. Bontempi, D. Raimondi, and K. Determe. On-board unit big data: Short-term traffic forecasting in urban transportation networks. 2020 IEEE 7th International Conference on Data Science and Advanced Analytics (DSAA), 2020, pp. 569-578, doi:10.1109/DSAA49011.2020.00072. 2020
* D. Raimondi, J. Simm, A. Arany, P. Fariselli, I. Cleynen, Y. Moreau, An interpretable low-complexity machine learning framework for robust exome-based in-silico diagnosis of Crohn’s disease patients, NAR Genomics and Bioinformatics, Volume 2, Issue 1, March 2020, lqaa011, https://doi.org/10.1093/nargab/lqaa011
* D. Raimondi, G. Orlando, P. Fariselli, and Y. Moreau. Insight into the protein solubility driving forces with neural attention. PLoS Computational Biology, 16(4), 2020. * D. Raimondi, G. Orlando, W.F. Vranken, and Y. Moreau. Exploring the limitations of biophysical propensity scales coupled with machine learning for protein sequence analysis. Scientific Reports, 9(1), 2019.
* G. Orlando, D. Raimondi, F. Tabaro, F. Codicè, Y. Moreau, and W.F. Vranken. Computational identification of prion-like rna-binding proteins that form liquid phase-separated condensates. Bioinformatics, 35(22):4617–4623, 2019.
* G. Orlando, D. Raimondi, and W. F. Vranken. Auto-encoding nmr chemical shifts from their native vector space to a residue-level biophysical index. Nature Communications, 10(1), 2019.
* D. Raimondi, G. Orlando, Y. Moreau, and W.F. Vranken. Ultra-fast global homology detection with discrete cosine transform and dynamic time warping. Bioinformatics, 34(18):3118–3125, 2018.
* D. Raimondi, G. Orlando, F. Tabaro, T. Lenaerts, M. Rooman, Y. Moreau, and W.F. Vranken. Large-scale in-silico statistical mutagenesis analysis sheds light on the deleteriousness landscape of the human proteome. Scientific Reports, 8(1), 2018.
* D. Raimondi, I. Tanyalcin, J.S.D. Fertè, A. Gazzo, G. Orlando, T. Lenaerts, M. Rooman, and W. Vranken. Deogen2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Research, 45(W1):W201–W206, 2017.
* G. Orlando, D. Raimondi, T. Khan, T. Lenaerts, and W.F. Vranken. Svm-dependent pairwise hmm: An application to protein pairwise alignments. Bioinformatics, 33(24):3902–3908, 2017.
* A. Gazzo, D. Raimondi, D. Daneels, Y. Moreau, G. Smits, S. Van Dooren, and T. Lenaerts. Understanding mutational effects in digenic diseases. Nucleic Acids Research, 45(15), 2017.
* D. Raimondi, G. Orlando, R. Pancsa, T. Khan, and W.F. Vranken. Exploring the sequence-based prediction of folding initiation sites in proteins. Scientific Reports, 7(1), 2017.
* D. Raimondi, G. Orlando, J. Messens, and W.F. Vranken. Investigating the molecular mechanisms behind uncharacterized cysteine losses from prediction of their oxidation state. Human Mutation, 38(1):86–94, 2017.
* D. Raimondi, A.M. Gazzo, M. Rooman, T. Lenaerts, and W.F. Vranken. Multilevel biological characterization of exomic variants at the protein level significantly improves the identification of their deleterious effects. Bioinformatics, 32(12):1797–1804, 2016.
* R. Pancsa, D. Raimondi, E. Cilia, and W. Vranken. Early folding events, local interactions, and conservation of protein backbone rigidity. Biophysical Journal, 110(3):572–583, 2016.
* G. Orlando, D. Raimondi, and W.F. Vranken. Observation selection bias in contact prediction and its implications for structural bioinformatics. Scientific Reports, 6, 2016.
* D. Raimondi, G. Orlando, and W.F. Vranken. An evolutionary view on disulfide bond connectivities prediction using phylogenetic trees and a simple cysteine mutation model. PLoS ONE, 10(7), 2015.
* D. Raimondi, G. Orlando, and W.F. Vranken. Clustering-based model of cysteine co-evolution improves disulfide bond connectivity prediction and reduces homologous sequence requirements. Bioinformatics, 31(8):1219–1225, 2015.
* M.J. Skwark, D. Raimondi, M. Michel, and A. Elofsson. Improved contact predictions using the recognition of protein like contact patterns. PloS Computational Biology, 10(11), 2014.


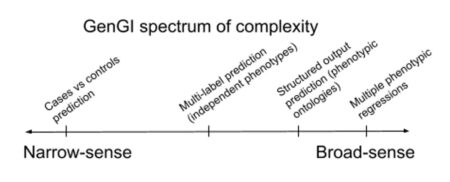 cases versus controls binary discrimination, while in the broad sense the goal is to predict multiple phenotypes for each sample. The papers on end-to-end GI that I have recently published 8,9,10,11 are the embryos from which I will expand this project. In particular, our Crohn’s disease (CD) case-control predictors8,9,10,11 belonged to the narrow sense GenGI, while Galiana, the multi-phenotypic prediction of A. thaliana samples8 is at the broad end of the spectrum, since it involved predicting hundreds of real-valued phenotypes, such as root length, flowering time, seed dormancy, etc. These papers show that the GenGI approach I propose can be a reality, and it already provides promising preliminary results. They contain in nuce the core aspects that I will develop to a full-fledged computational framework during the 5 years of my current CPJ position at IGMM.
cases versus controls binary discrimination, while in the broad sense the goal is to predict multiple phenotypes for each sample. The papers on end-to-end GI that I have recently published 8,9,10,11 are the embryos from which I will expand this project. In particular, our Crohn’s disease (CD) case-control predictors8,9,10,11 belonged to the narrow sense GenGI, while Galiana, the multi-phenotypic prediction of A. thaliana samples8 is at the broad end of the spectrum, since it involved predicting hundreds of real-valued phenotypes, such as root length, flowering time, seed dormancy, etc. These papers show that the GenGI approach I propose can be a reality, and it already provides promising preliminary results. They contain in nuce the core aspects that I will develop to a full-fledged computational framework during the 5 years of my current CPJ position at IGMM.


