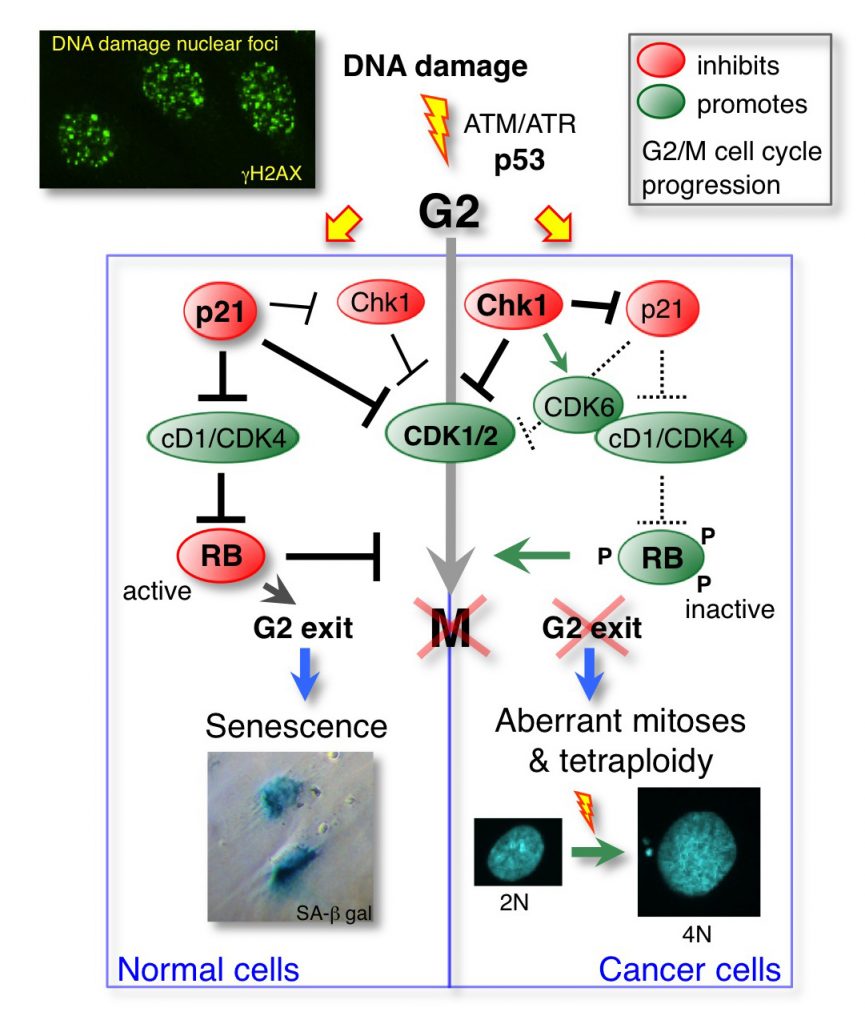
Cellular senescence is an irreversible proliferation withdrawal that can be initiated after DNA damage-induced cell cycle arrest in G2 phase to prevent genomic instability caused by aberrant mitoses. Its onset requires p53 (TP53) and retinoblastoma protein (RB1) family tumour suppressors, but how a temporary cell cycle arrest is converted into a permanent one remains unknown. We show that a previously unrecognised balance between the p53-regulated cyclin-dependent kinase (CDK) inhibitor p21 and the checkpoint kinase Chk1 controls cyclin D1–CDK activity during G2 arrest. In non-transformed cells, p21 activates RB in G2 by inhibiting cyclin D1 complexed with CDK2 or CDK4. The resulting G2 exit, which precedes the appearance of senescence markers, is associated with a mitotic bypass, Chk1 downregulation and reduction in the number of DNA damage foci. In p53/RB-proficient cancer cells, a compromised G2 exit correlates with sustained Chk1 activity, delayed p21 induction, untimely cyclin E1 re-expression and genome reduplication. Conversely, Chk1 depletion promotes senescence by inducing p21 binding to cyclin D1– and cyclin E1–CDK complexes and downregulating cyclin D partner CDK6, whereas knockdown of the checkpoint kinase Chk2 increases RB phosphorylation and delays G2 exit. In conclusion, p21 and Chk2 oppose Chk1 to maintain RB activity, thus promoting the onset of senescence induced by DNA damage in G2.
Lossaint, G*., Horvat, A*., Gire, V., Bačević, K., Mrouj, K., Charrier-Savournin, F., Georget, V., Fisher, D., and Dulić, V. (2022) Reciprocal regulation of p21 and Chk1 controls the Cyclin D1-RB pathway to mediate senescence onset after DNA damage-induced G2 arrest. J. Cell Sci. 135, jcs259114. doi: 10.1242/jcs.259114