

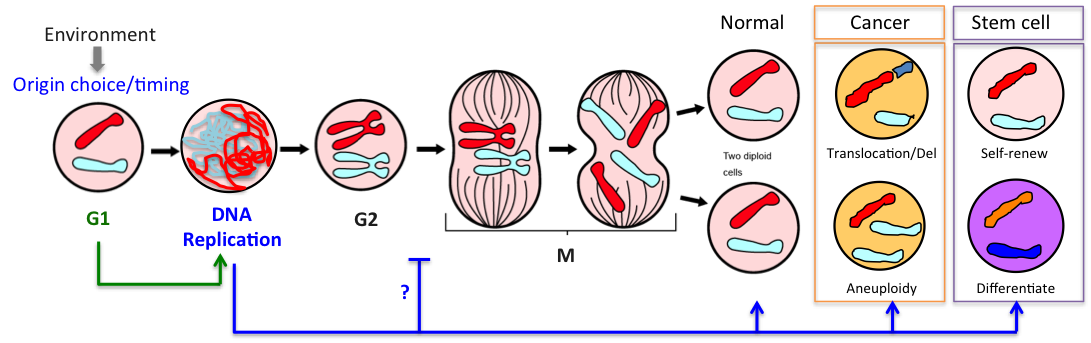
All eukaryotic cells duplicate their genome using cell-type specific spatio-temporal replication programs, which are still poorly understood in spite of their key roles in genome stability and chromatin inheritance. We use yeast and mammalian cells to understand how these programs are established, how they contribute to cell identity and how their perturbation leads to loss of genome integrity. Since replication origin firing is under tight cell cycle control, we use biological models that depart naturally (stem cells, post-quiescent cells, meiotic cells) or genetically (defined mutants, cancer cell lines) from the standard cell cycle to uncover specific replication patterns and to study their involvement in genome stability and cell identity.
A key but often overlooked property of DNA replication is its inherent stochastic nature, which calls for single-cell or single-molecule analysis. We introduced new methods, such as single-molecule analysis of DNA replication by DNA combing, to better assess chromosome replication dynamics in individual cells, which led us to uncover specific replication patterns in cancer cells and stem cells.
1.G1: the programming phase
G1 is the key period of the cell cycle when chromosomes adopt their interphase structure, when replication origins are licensed and their timing programmed. Licensing, i.e assembly of pre-replication complexes (pre-RC) onto origins, requires CDK activity to be low. We showed previously that yeast cells lacking the G1 CDK inhibitor Sic1 (ortholog of p27) license fewer origins, have an extended S phase and frequently rearrange their chromosomes (Lengronne & Schwob, 2002). Incidentally cancer cells also have a truncated G1 phase and high rates of chromosome instability. Thus G1 control is key for S-phase dynamics and genome stability.
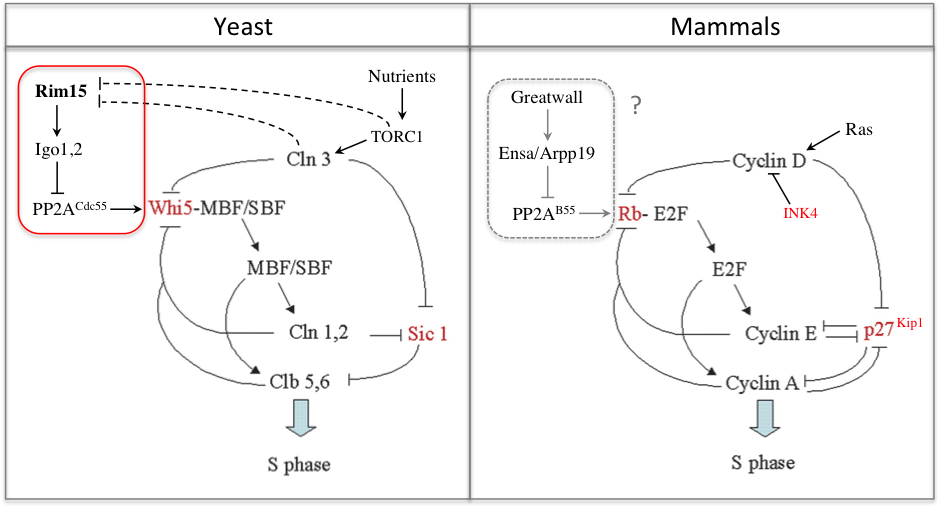
In a recent study we found that the G1 restriction point (called START in yeast) is controlled by the Rim15 kinase (Greatwall in metazoans), which inhibits the PP2ACdc55 phosphatase counteracting Whi5 phosphorylation by Cln3-Cdk1 upon nutrient-limiting conditions (Talarek et al., 2017). Rim15 activation promotes START when Cln3 levels are low and might explain why cells grown in poor nutrients are actually smaller, not larger, than cells grown in rich medium. Given the similarity of G1/S control in yeast and metazoans, one can wonder if Gwl kinase, which is frequently overexpressed in cancer, might not control Rb phosphorylation and the restriction point in mammals.
2.S: the execution phase
DNA synthesis begins from numerous replication origins that are activated throughout S phase following cell type specific spatio-temporal programs. Since each cell uses a different combination of origins, we devised IdU-CldU pulse labelling protocols and single DNA molecule visualization methods to determine the dynamics of chromosome replication at the single cell level, without averaging data over the entire cell population.

Several parameters are determined: fork velocity (FV), fork processivity (FP), inter-origin distances (IODs) and global instant fork density (GIFD). We recently found that the latter parameter changes significantly in cancer cells (Coulon et al., in prep) and in stem cells (Bialic et al., in prep).
To see the incidence of these changes on S-phase duration (SPD), we developed a robust method, based on dual EdU-BrdU pulse separated by increasing thymidine chases, to measure SPD from asynchronous cells. This led to the observation that SPD is significantly extended in all cancer cell lines tested so far. Mechanistic studies are underway to pinpoint the origin and consequences of SPD extension in cancer cells.
3.M: the money time phase
Mitosis is the time when properly replicated chromosomes are segregated in daughter cells. But it is also the time when cells pay the price if DNA replication dynamics has been altered.
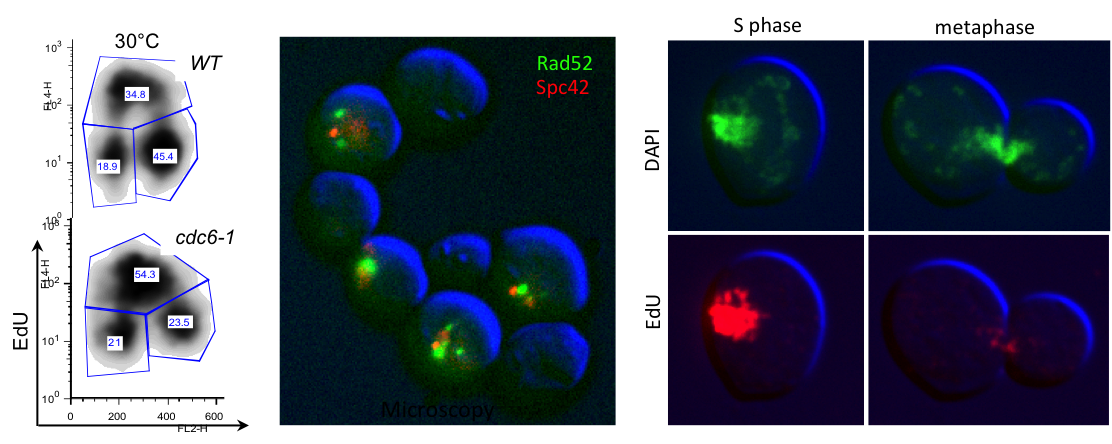
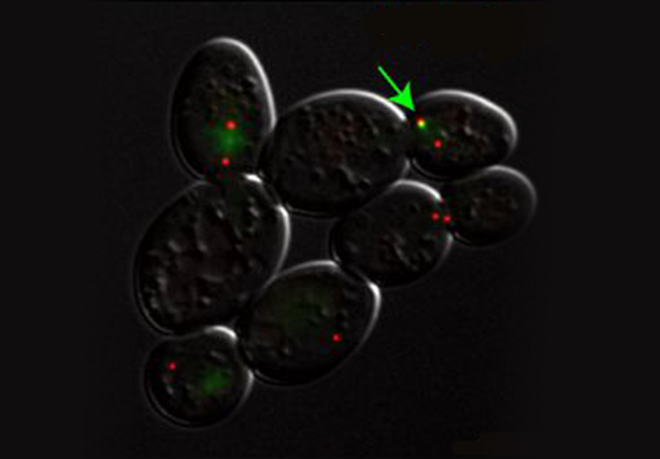
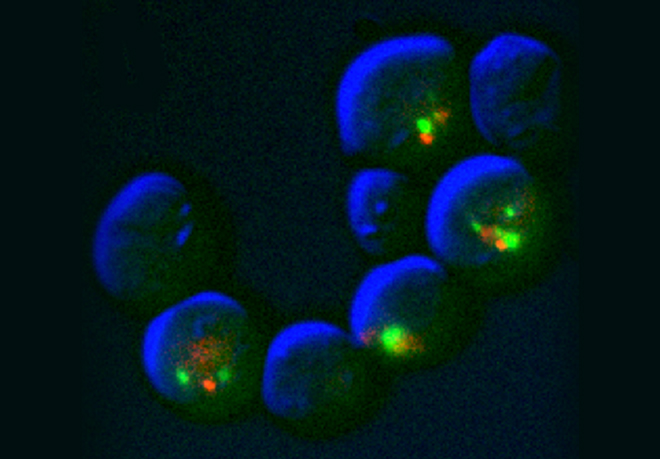
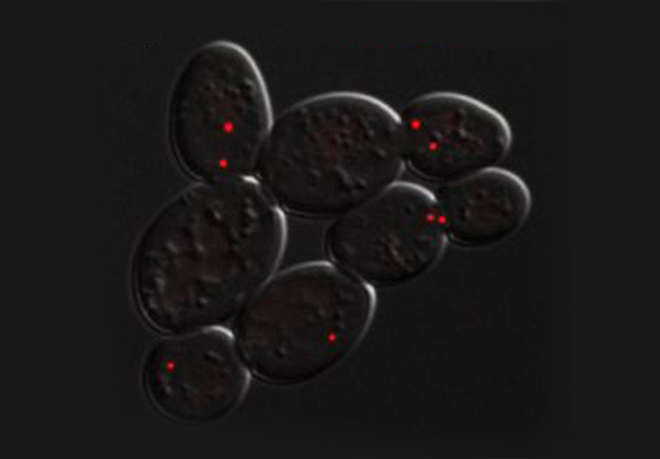
For example when S phase is extended by decreasing origin licensing in yeast (cdc6-1 ts mutant at semi-permissive T°), cells form Rad52 recombination foci at the time they enter mitosis with incompletely replicated chromosomes and finish the job in an error-prone manner in metaphase (Montecchi et al., in prep). We identified the mechanism for this switch from normal to break-induced replication (BIR) in yeast, and are now testing if a similar phenomenon takes place at sites of very late replication in cancer cells to generate chromosome instability.
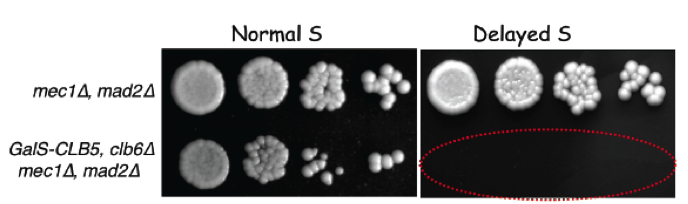
We also showed that yeast cells entering S phase later than normal delay mitotic entry using both the S-phase (Mec1, ATR) and spindle assembly (Mad2, SAC) checkpoints, which become essential when S phase is delayed (Magiera et al., 2014). These cells also rely on Bub1 and Kar3 for proper chromosome bi-orientation, underlining the importance of chromosome replication dynamics for accurate chromosome segregation in mitosis.









