

Our research focuses on understanding how higher-order 3D chromatin organization is involved in the control of gene expression in mammals in both physiological and pathological contexts.
In physiological contexts, through the development of original experimental techniques and modeling tools, our team is unravelling properties of chromatin organization at two important levels: Topologically-Associating Domains (TADs) (Axis 1) and nuclear compartments linked to nuclear bodies.
In pathological contexts, we are investigating the regulation of the SNCA gene encoding the α-synuclein, a major protein of the Parkinson disease.
1.
Coordination of mammalian gene expression within TADs
Our group contributed to the development of the Chromosome Conformation Capture (3C) technologies by improving the sensitivity of the 3C assay (3C-qPCR method) (Hagège et al., Nature Protocols 2, 1722, 2007; Rebouissou et al., Meth. Mol. Biol. 2532, 187-197, 2022). Using this method, we showed that gene-rich loci display modulated contact frequencies (Court et al., Genome Biol. 12, R42, 2011). After initiating an interdisciplinary project in association with Annick Lesne, physicist, we showed that this modulation can be described by polymer models as if the mammalian chromatin was folded statistically into a helix shape (statistical helix model) (Ea et al., Genes 6, 734-750, 2015). We then showed that, due to their different size, distinct principles of polymer physics govern chromatin dynamics in mouse and Drosophila topological domains (Ea et al., BMC Genomics 16, 607, 2015; collab. with G. Cavalli, Montpellier).
More recently, we showed that some endogenous retroviral (ERV) sequences act in the mouse genome as putative enhancers controlling endogenous gene expression through long-range chromatin interactions controlled by HP1 (Heterochromatin Protein 1) (Calvet et al., Cells 11, 2392, 2022; collab. with F. Cammas, IRCM, Montpellier).
Finally, in an in-silico study, using genome-wide association studies (GWAS), we showed that, for a fraction of human diseases, diseases-associated non-coding Single Nucleotide Polymorphisms (SNPs) are preferentially located in TAD borders (Jablonski et al., Human Genomics, 16, 2, 2022; collab. with M.T. Hütt, Bremen, Germany). Remarkably, cancers are relatively more frequent among these diseases and, with the support of the ARC Foundation, we are now characterizing the higher-order 3D chromatin organization of one such paradigmatic locus associated with lung cancer risk.
2.
Nuclear bodies and higher-order chromatin organization in mammals
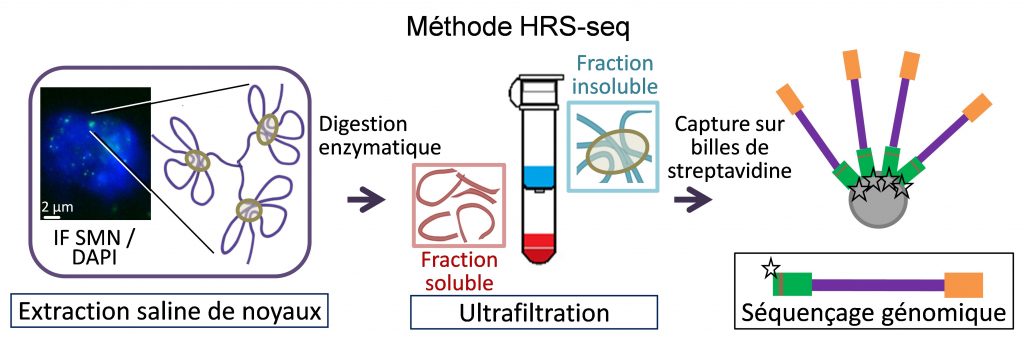
Nuclear bodies are membrane-less sub-nuclear organelles that essentially behave as liquid-like droplets. Many of them assemble in vivo through phase separation processes and play an essential role in coordinating efficient expression of some genes by confining together specific chromatin regions dispersed throughout the genome (Lesne et al., Genes 10, 1049, 2019). We developed a novel experimental approach (HRS-seq method) for genome-wide analyses of sequences associated with large ribonucleoproteic (RNP) complexes, including nuclear bodies. Using mouse Embryonic Stem (ES) cells, we showed that the active chromosomal compartment is associated with such large RNP complexes (Baudement et al., Genome Res. 28, 1733-1746, 2018; collab. with L. Journot, IGF, Montpellier and J. Mozziconacci, MNHN, Paris).
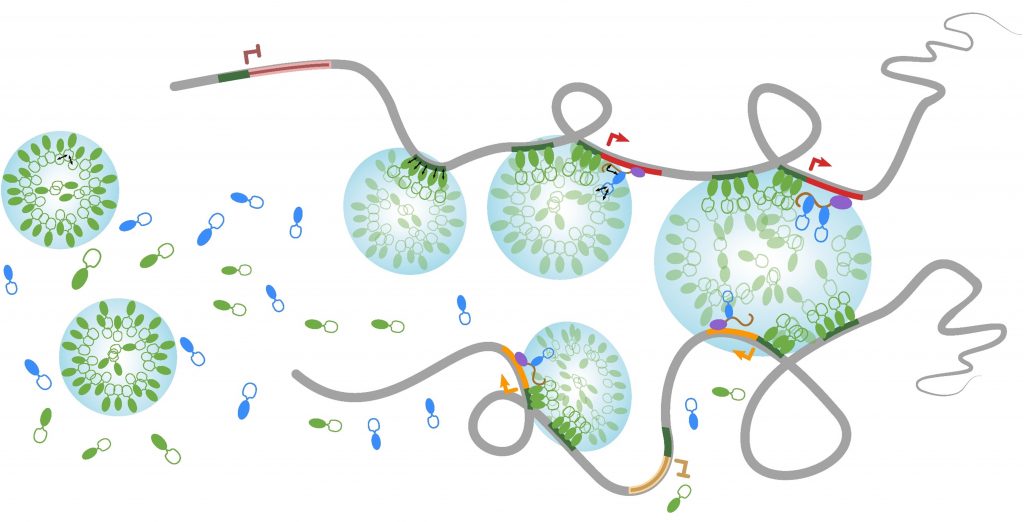
We are now exploring the role that these nuclear bodies play in higher-order chromatin organization and in the control of gene expression, during cell differentiation or in some pathological situations. In order to more accurately identify genes associated with a specific class of nuclear bodies, we are adapting the HRS-seq method by acting on the physical parameters of phase separation (ARGECOR 80Prime project, supported by the CNRS interdisciplinary mission - MITI).
3.
Transcriptional regulation of α-synuclein expression in Parkinson's disease
Parkinson's disease, whose motor symptoms are mainly due to the selective disappearance of the dopaminergic neurons of the Substantia Nigra pars compacta (SNpc), is the second most common neurodegenerative disease in the Human. The majority of cases result from a combination of genetic and environmental factors. The SNCA gene, which is encoding for the α-synuclein protein, a neuronal protein present at presynaptic terminals and playing a role in the release of neurotransmitters, is the first gene to have been associated with Parkinson's disease. Even a limited increase in the expression of wild-type α-synuclein can cause familial or sporadic forms of Parkinson's disease. Several single nucleotide polymorphisms (SNPs) at the SNCA locus, identified in genome-wide association studies (GWAS) as being strongly associated with an increased risk of Parkinson's disease, correlate with higher levels of α-synuclein in patients. Therefore, reducing the expression of α-synuclein represents an interesting therapeutic strategy. To achieve this, we need to have a thorough understanding of the molecular mechanisms involved in the transcriptional regulation of SNCA in normal conditions, but especially in pathological conditions.
With the support of the Fondation pour la Recherche Médicale (FRM), we are investigating the mechanisms of SNCA gene activation in normal and pathological conditions. We are using cell models representing sporadic forms (LUHMES cells grown in 3D, differentiated into dopaminergic neurons and treated with MPP+) or familial forms (organoids derived from induced pluripotent cells carrying the α-synuclein p.A53T mutation) of Parkinson's disease. Using these models, we are characterizing the key genomic regulatory elements and transcription factors responsible for the pathological induction of SNCA. To this aim, in collaboration with Solange Desagher (IRIM, Montpellier), Franck Court (GReD, Clermont-Ferrand) and Philippe Ravassard (ICM, Paris), we are combining approaches based on 3D chromatin architecture (HRS, 4Cseq) with available GWAS data, proteomics and genome editing using CRISPR-Cas9.





